Sterile Pharmaceutical Products 5z6m2i
This document was ed by and they confirmed that they have the permission to share it. If you are author or own the copyright of this book, please report to us by using this report form. Report 2z6p3t
Overview 5o1f4z
& View Sterile Pharmaceutical Products as PDF for free.
More details 6z3438
- Words: 4,164
- Pages: 62
]
|
|
!" # To review basic GMP requirements in the manufacture of sterile pharmaceutical products To review air classifications for activities related to the manufacture of sterile products To review the different types of sterilization methods To review quality assurance aspects in the manufacture and control of sterile products To consider current issues applicable in your country |
|
$ ]dditional rather than replacement Specific points relating to minimizing risks of contamination ± microbiological ± particulate matter ± pyrogen
|
|
% Production in clean areas ]ppropriate standard of cleanliness Filtered air supplied ]irlocks for entry ± Personnel and/or equipment ± Materials Separate areas for operations ± component preparation (containers and closures) ± product preparation, filling, sterilization, etc. |
|
÷ ÷ ÷
uesign ± ]void unnecessary entry of supervisors and control personnel ± Operations observed from outside In clean areas, all exposed surfaces: ± Smooth, impervious, unbroken ± Minimize shedding and accumulation of particles, microorganisms ± Permit cleaning and disinfection ± No uncleanable recesses, ledges, shelves, cupboards, equipment ± Sliding doors undesirable ± False ceilings sealed M ÷ M |
|
In clean areas, all exposed surfaces (2): ± Proper installation of pipes and ducts, no recesses, no unsealed openings ± Sinks and drains avoided, and excluded in Grade ] and B areas ÿ ÿ
ÿ ÿ
M
|
|
hanging rooms ± uesigned as airlocks ± Effective flushing with filtered air ± Separate rooms for entry and exit desirable ± Hand washing facilities ± Interlocking system for doors ± Visual and/or audible warning system Use filtered air supply to maintain pressure cascade Pressure differential approximately 10 to 15 Pascales Zone of greatest risk ± immediate environment |
|
M M M
Pathogenic, highly toxic, radioactive materials Pressure cascade may be different uecontamination procedures ± air, equipment, garments Qualification including airflow patterns ± No risk to the product Warning system to indicate failure in air supply Pressure indicators ± results regularly recorded Restricted access ± e.g. use of barriers |
|!
M M M ÷
&$ onveyer belts Effective sterilization of equipment Maintenance and repairs from outside the clean area ± If taken apart, resterilized before use ± Use clean instruments and tools Planned maintenance, validation and monitoring ± Equipment, air filtration systems, sterilizers, water ÷ ÷ ÷ treatment systems |
|"
&$ Water treatment plants and distribution system ± uesign, construction, maintenance ± Operation and design capacity ± Testing programme
Water for Injection (WFI) ± Produced, stored, distributed ± prevention of growth of microorganisms ± onstant circulation at temperature above 70, or not more than 4 degrees elsius |
|
÷
& # ' ( !
]ir samples Surface swabs Personnel swabs
|
|
& # ' (( Particulate matter uifferential pressures ]ir changes, airflow patterns lean-up time/recovery Filter integrity Temperature and relative humidity ]irflow velocity |
|
Frequent, thorough cleaning of areas necessary Written programme Regular monitoring to detect resistant strains of microorganisms hemical disinfection Monitoring of disinfectants and detergents uilutions ± lean containers, stored for defined periods of time ± Sterilized before use, when used in Grade ] or B areas |
|
¦ ÷ ¦
Monitoring of clean areas Monitoring of personnel and surfaces after critical operations Frequent monitoring in areas where aseptic operations are carried out ± Settle plates, volumetric air samples, surface sampling (swabs and plates) ± Sampling methods should not contaminate the area Results considered when batch release is done |
|
¦¦
Gimits of detection established ]lert and action, and monitoring trends of air quality
¦
Table 1. Gimits for microbial contamination (Information only) „
„
Grade
Air sample
Settle plates
plates
Glove print
(CFU/m3)
(90mm diameter)
(55mm diameter)
(5 fingers)
(CFU/4hours)
(CFU/plate)
(CFU/glove)
A
<3
<3
<3
<3
B
10
5
5
5
C
100
50
25
-
D
200
100
50
-
„ |
|
Minimum number of personnel in clean areas ± Especially during aseptic processing Inspections and controls from outside Training to all including cleaning and maintenance staff ± Initial and regular ± Manufacturing, hygiene, microbiology Special cases ± Supervision in case of outside staff Î ÷ Î ¦ ± uecontamination procedures (e.g. staff who worked with animal tissue materials) |
|
High standards of hygiene and cleanliness Periodic health checks No shedding of particles No introduction of microbiological hazards No outdoor clothing hanging and washing procedure No watches, jewellery and cosmetics
|
|
Î Î
lothing of appropriate quality: ± Grade u hair, beard, moustache covered Protective clothing and shoes ± Grade Hair, beard, moustache covered Single or 2-piece suit (covering wrists, high neck), shoes no fibres to be shed Î ± Grade ] and B Headgear, beard and moustache covered, masks, gloves No shedding of fibres, and retain particles shed by operators |
|!
Outdoor clothing not in change rooms leading to grade B and rooms hange at every working session, or once a day (if ive data) hange gloves and masks at every working session uisinfect gloves during operations Washing of garments ± separate laundry facility No damage, and according to validated procedures |
|"
Î Î Î M
) åou are asked to visit a factory producing the following
product lines: ± Injections in ampoules and vials, including insulin, vaccines and heat-stable pharmaceuticals ± Sterile eye ointment
uescribe the type of facility you would expect to find Gist the typical rooms, their purpose and air classification |
|
!( Poor design of the building Poor design of the systems, e.g. water, HV] Flow of personnel Flow of material No validation or qualification Old facilities not complying with current requirements
|
|
!(* + Particulate levels/microorganisms uifferential pressures ]ir changes Temperature/humidity
|
|
, -
Terminally sterilized ± prepared, filled and sterilized
]septic preparation ± some or all stages ÷¦ |
|
lassification of clean areas Manufacturing operation in an appropriate environment cleanliness level Minimize risks ± particulate and microbiological contamination ± product and material Meet classification "at rest" ± (i.e. "completed installation, equipment installed and operating, but no operating personnel present"). ÷ |
|
For sterile pharmaceutical preparations: Grade ] ± Gocal zone, high risk operations, e.g. filling, aseptic connections ± Usually Uu]F systems used Grade B ± Background environment to grade ] (in case of aseptic preparation and filling) Grade and Grade u ± lean areas for less critical operations |
|
÷
] !
. . /
0./
1.
B
1.
C D
1. 1.
. . / 1. 1.
1.
0./
„ ¦÷
|
|
To reach Grade B, and u, the number of air changes should be appropriate to the size of the area, number of personnel, equipment present Minimum of 20 air changes per hour lean up time about 15-20 minutes Good airflow pattern in the area HEP]-filtered air Suitable methods to determine particulate matter and micro± e.g. EU, ISO, Japan, US] ÷ |
|
ontrol particulate during operation Monitoring during operation ]lert and action limits for particulate and micro ]ction taken when exceeded ]rea grades should be proven (e.g. validation runs, media fills, environment, time limits ± based on microbiological contamination/bioburden found)
¦
|
| !
]
23
4
4
5
A
1 .
.
A
6
1 .
.
6
. .
7
8
.
9
8
„ ÷
|
| "
Minimise contamination ± all stages including before sterilization and during processing No unsuitable materials, e.g. live microbiological organisms Minimize activities ± staff movement controlled and methodical ± avoid shedding of particles Temperature and humidity comfortable ontainers and materials in the area |
|
÷ ÷ ÷
Validation ± should not compromise the processes ]septic process validation: Sterile media fill (³broth fills´) ± Simulate actual operation ± intimate as closely as possible ± Simulate worst expected condition ± Use appropriate medium/media ± Sufficient number of units - e.g. equal to batch size (small batches) acceptable limit investigations ± Revalidation: periodic and after change New processing procedures validated ± Revalidation after significant changes ± ]nd regular intervals |
|
÷ ÷Î Î
Water sources, water treatment systems and treated water Monitored regularly ± hemicals ± Biological contamination ± Endotoxin Water specification Records of results and action taken |
|
÷M
omponents, bulk product containers and equipment ± fibre generation ± no recontamination after final cleaning ± stage properly identified ± sterilized when used in aseptic areas Used in clean areas, ed through double-ended sterilizers or use triple wrapping ¦ Gas used to purge solution or blanket a product ± ed through a sterilizing filter |
|
Bioburden monitored ± Products: Before sterilization ± Working limits established ± Solutions to be filtered before filling (especially GVP) ± Pressure release outlets ± hydrophobic microbiological air filters Starting materials ± microbiological contamination should be minimal Monitored as per specification |
|
¦
#-% !: $ Washing and drying and sterilization; and sterilization and use ± ]s short as possible ± Time limit validated #- Start of preparation of solution and sterilization (filtration) ± ]s short as possible ± Maximum time set for each product ¦ |
|
onsidering the same factory as in the previous group session, discuss the process of sterilization. Gist all the items that will need to be sterilized (and indicate the choice of sterilization process). What are the key features you should find in each sterilization situation? uiscuss the relevance, need, and the extent of qualification and validation required.
|
|
!( ]utoclave - no pressure gauge ]utoclave - no temperature recorder ]utoclave - superheated steam lean room - pressure differentials Exposure for settle plates Interlocks turned off Rusty Gaminar airflow cabinets HEP] filters not checked regularly |
|
; Methods of sterilization: ± ± ± ±
Moist or dry heat Irradiation (ionizing radiation) Sterilizing gaseous agents (e.g. ethylene oxide) Filtration with subsequent aseptic filling
Whenever possible: Terminal sterilization by heat in their final container - method of choice ÷ |
|!
; Validation ± ]ll sterilization processes ± Special attention when non-pharmacopoeia methods are used ± Non-aqueous or oily solutions Before the method is adopted ± its suitability and efficacy demonstrated with desired conditions: ± ]ll parts of the load ± Each type of load ± Physical measurements and biological indicators (where appropriate) ± Verified at least annually and after change ± Records maintained |
|"
; For effective sterilization: Whole of the material subjected to the treatment Biological indicators: ]dditional method of monitoring Storage and use, quality checked through positive control Risk of contamination |
|
; uifferentiation between sterilized and not-yet-sterilized products Each basket/tray or other carrier, properly labelled ± Name of material ± Batch number ± Sterilization status
Use of autoclave tape Sterilization records for each run ± approved as part of Î M the batch release procedure |
|
; Sterilization by heat Sterilization by moist heat Sterilization by dry heat Sterilization by radiation Sterilization by gases and fumigants |
|
; Sterilization by heat Recording of each cycle, e.g. time and temperature chart ± Temperature: validated coolest part ± heck from second independent probe ± ]dditional chemical or biological indicators Heating phase: Sufficient time for the whole load ± uetermined for each load ooling phase: ]fter sterilization cycle ± Precautions to prevent contamination ± Sterilized cooling fluid/gas |
|
¦
; Sterilization by moist heat (heating in an autoclave) Water-wetable materials only, and aqueous formulations Temperature, time and pressure monitored Temperature recorder independent of the controller Independent temperature indicator urain ± temperature recorded from this position Regular leak test when vacuum is part of the cycle Material allows for removal of air and penetration of steam ]ll parts of the load in with steam Quality of the steam ± no contamination |
|
; Sterilization by dry heat For non-aqueous liquids, dry powders ]ir circulation in the chamber Positive pressure in chamber to prevent entry of non-sterile air HEP] filtered air supplied When removing pyrogens, challenge tests ± validation (using endotoxins) |
|
; Sterilization by radiation Suitable for heat-sensitive materials and products ± confirm suitability of method for material ± ultraviolet irradiation not acceptable ontracting service ± ensure validation status, responsibilities Measurement of dose during procedure uosimeters independent of dose rate ± quantitative measurement ± number, location and calibration time-limit Biological indicators only as additional control Radiation sensitive colour discs |
|
Î ÷
; Sterilization by radiation (2)
Information forms part of the batch record Validation to cover effects of variation in density of
packages
Handling procedures to prevent misidentification of irradiated and non-irradiated materials Each package to have a radiation-sensitive indicator Total radiation dose istered within a predetermined period of time ÷ ÷¦ |
|
; Sterilization by gases and fumigants Only when no other method is suitable e.g. ethylene oxide, hydrogen peroxide vapour Validation: ]lso prove the gas has no damaging effect on product Time and conditions for degassing (specified limits) - residue uirect with microbial cells essential ± Nature and quantity of packaging materials Humidity and temperature equilibrium Monitoring of each cycle with biological indicators ± time, pressure ± temperature, humidity and gas concentration |
|!
÷
; Sterilization by gases and fumigants (2)
Post-sterilization storage ± controlled manner ± Ventilated conditions ± uefined limit of residual gas ± Validated process
Safety and toxicity issues ÷ |
|"
;
u
„
|
|
„
;
]
!
„
Î M
|
|
] ; ! ]septic processing Objective is to maintain the sterility of a product, assembled from sterile components Operating conditions so as to prevent microbial contamination What do you think are the aspects that require careful attention? ÷ |
|
] ; ! ]septic processing (2) areful attention to: Environment Personnel ritical surfaces ontainer/closure sterilization Transfer procedures Maximum holding period before filling |
|
¦
]
u
„
÷ ÷÷ ÷
|
|
] ] " # $
] " # $ ] " # $ ] " # $ ] " # $
%
|
"& ' $
" $ (
( ) *
„ |
÷ ÷¦
"+ # $
; ! Through a sterile filter of 0,22 µm or less, into previously sterilized containers ± remove bacteria and moulds ± not all viruses or mycoplasmas onsider complementing with some degree of heat treatment uouble filter layer or second filtration advisable, just before filling - no fibre shedding or asbestos filters Filter integrity testing immediately after use ± also before use if possible |
|
; !< *+ Validation to include ± Time taken to filter a known volume ± Pressure difference to be used across the filter
Significant differences to be noted and investigated, recorded in batch records Integrity of gas and air vent filters checked after use, other filters at appropriate intervals |
|
; !< * + Same filter not used for more than one working day, unless validated No filter interaction with product, e.g. ± removal of ingredients ± releasing substances into product
Î M |
|!
= % Samples for sterility testing should be representative From parts of the batch, most at risk ± ]septic filling ± at beginning and end of batch filling, and after interruptions ± Heat sterilized ± coolest part of the load Sterility of the batch ensured through validation ± Validated sterilization cycle ± Media fill ÷ Sterility test procedure as per pharmacopoeia, and validated for eachproduct
Batch processing records, sterility testing records, environmental records should be reviewed
|
|"
= % Endotoxin testing for injectable products ± Water for injection, intermediate and finished product
]lways for large volume infusion solutions Pharmacopoeia method, validated for each product Failure of the test ± investigation orrective action |
|
¦
< ontainers closed by means of validated methods Samples checked for integrity Maintenance of vacuum (where applicable) checked Parenteral products inspected individually Visual inspection under suitable and controlled conditions: ± illumination and background ± eyesight checks of operators ± allowed frequent breaks Other methods: ± validated, and equipment performance checked at intervals ± results recorded |
|
÷÷ ÷ ÷÷ ¦
onsidering the same factory as in the previous group sessions, devise a plan for monitoring of the facility. Gist the parameters to be tested, tests to be used, acceptance criteria and frequency of testing.
|
|
Related Documents c2h70

Sterile Pharmaceutical Products 5z6m2i
October 2019 40
Sterile Pharmaceutical Products 5z6m2i
October 2019 87
Sterile Pharmaceutical Products - Kennethe. Avis.pdf 181f4q
November 2021 0
Quality Control Of Sterile Products 5q5t36
December 2019 34
Packaging Components Of Pharmaceutical Products.! 2x41h
October 2019 33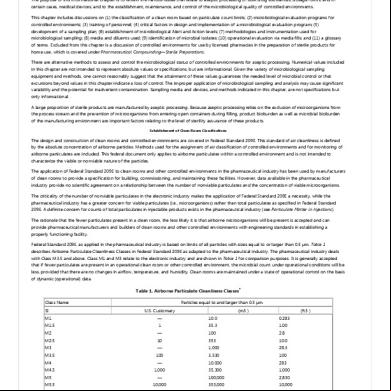
Clean Rooms And Controlled Areas (sterile Area Classification) _ Pharmaceutical Guidelines.pdf 4g4m4a
November 2019 74More Documents from "Taesup Byun" 105y
Sterile Pharmaceutical Products 5z6m2i
October 2019 40